Breast Cancer's Complexity Pushes Researchers to Look Beyond the Genome
Although breast cancer research has helped chart the course for molecularly targeted therapies in oncology, next-generation sequencing technologies have revealed a disease so highly complex and heterogeneous that translating these findings into clinically useful therapies has proved daunting.
Although breast cancer research has helped chart the course for molecularly targeted therapies in oncology, next-generation sequencing (NGS) technologies have revealed a disease so highly complex and heterogeneous that translating these findings into clinically useful therapies has proved daunting.
Indeed, genomic complexities uncovered thus far potentially impact current treatment paradigms because of an increased risk that clinicians are basing decisions on incomplete information. Meanwhile, this complexity limits the broad clinical applicability of any potentially druggable genomic events, affecting future therapeutic developments.
A Subtype-Specific View
Now, experts are turning increasingly toward newer technologies that will allow them to dissect the cellular outcomes of the many genomic alterations already uncovered, as well as those yet to be revealed. There is hope that shifting focus away from individual mutations and back to the cancer cell as a whole will help to bridge the gap between genomics and clinical action.Currently, clinicians base their treatment decisions for patients with breast cancer on disease histology and the expression of the estrogen receptor (ER), the progesterone receptor (PR) and the human epidermal growth factor receptor 2 (HER2), all of which have well-established roles as drivers of this disease.
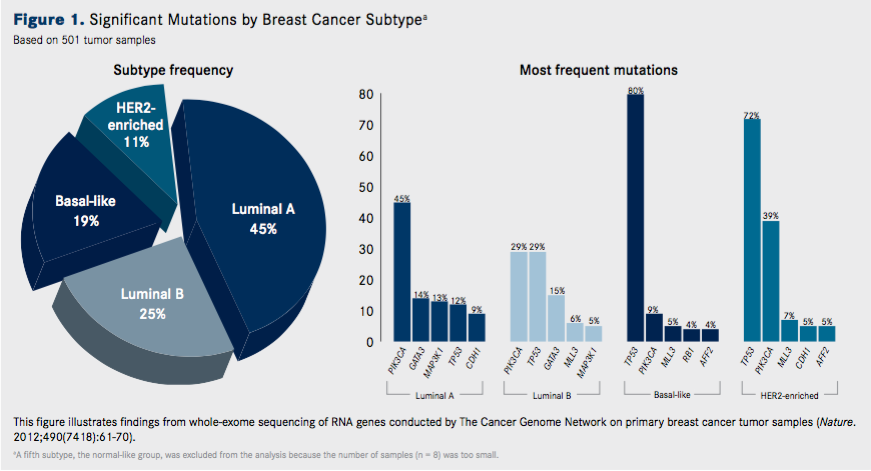
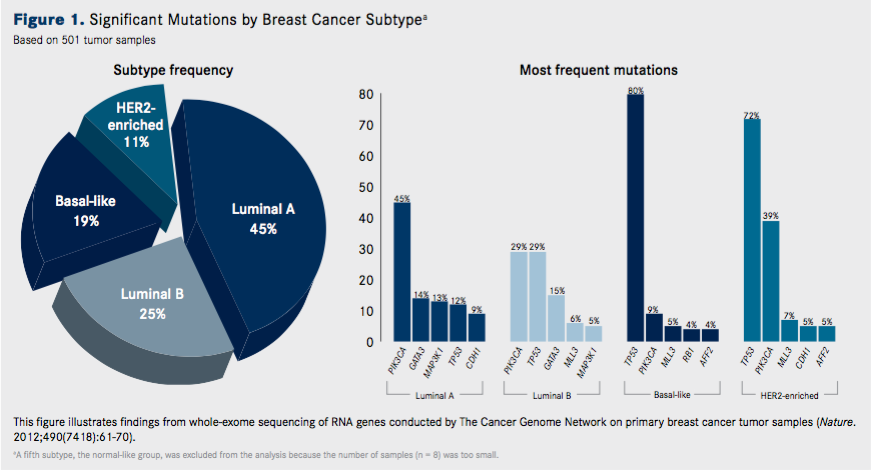
A genomic view of the tumors looks beyond that approach. Clinically speaking, there are at least 5 recognized gene expression—based subtypes of breast cancer: luminal A and luminal B, both of which express ER and PR; HER2-enriched tumors, which express the HER2 protein; basal-like tumors; and a normal breast-like subtype that remains ill-defined.
The luminal A and luminal B tumors are the most prevalent among the subtypes. Basal-like tumors, which represent the most heterogeneous subtype, overlap to a large extent with triple-negative breast cancers (TNBCs), defined by their lack of ER, PR, or HER2 expression. Around 75% of TNBCs fall into the basal subtype, while the remaining quarter are distributed across the other subtypes.
In 2012, a series of NGS studies fueled an explosion in the breast cancer genomics field. These studies began to piece together a more comprehensive picture of the genomic events driving breast cancer.
Substantial heterogeneity was observed not only between the different clinical subtypes, but also within them, as well as within individual patients and between the primary tumor and metastases.
Intrapatient heterogeneity was highlighted in a recent study in which single-nucleus genome sequencing was performed on 50 individual cancer cells. Within a single TNBC sample, there were seemingly 3 different cancers and no 2 cancer cells had the same genome.
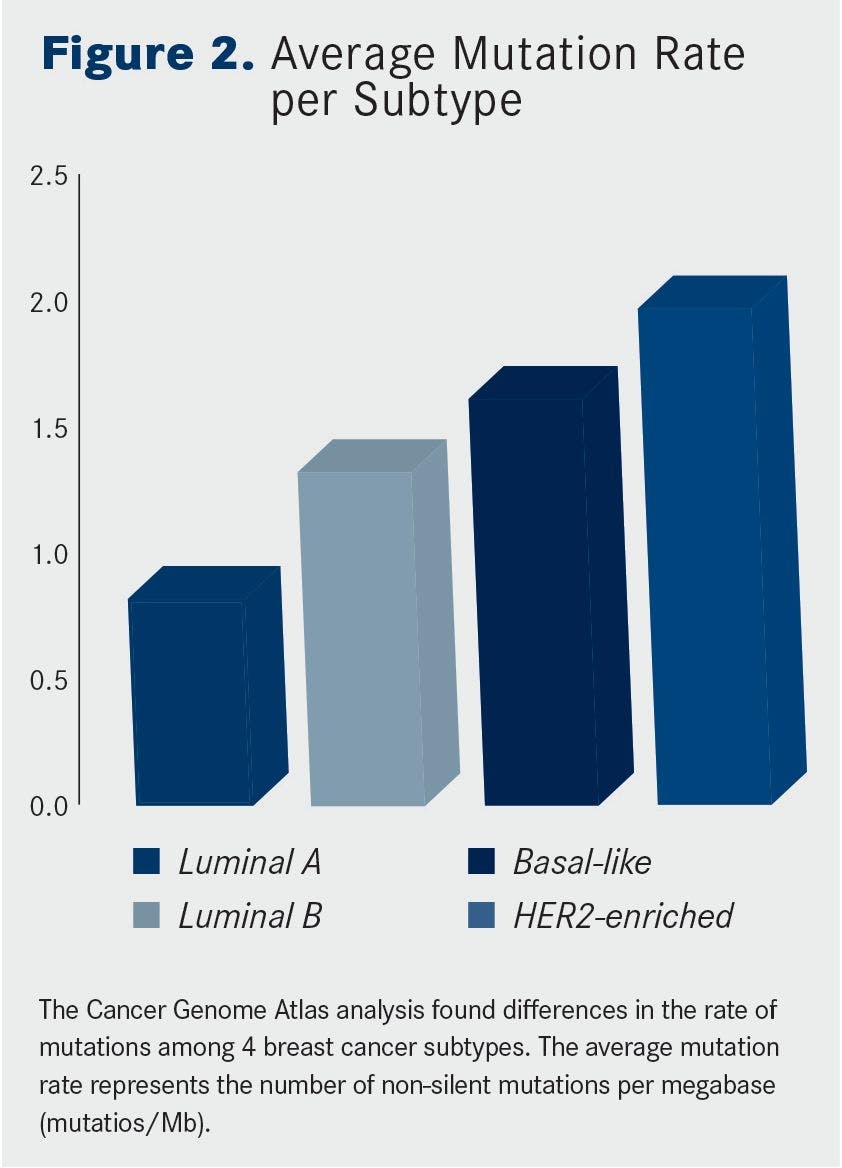
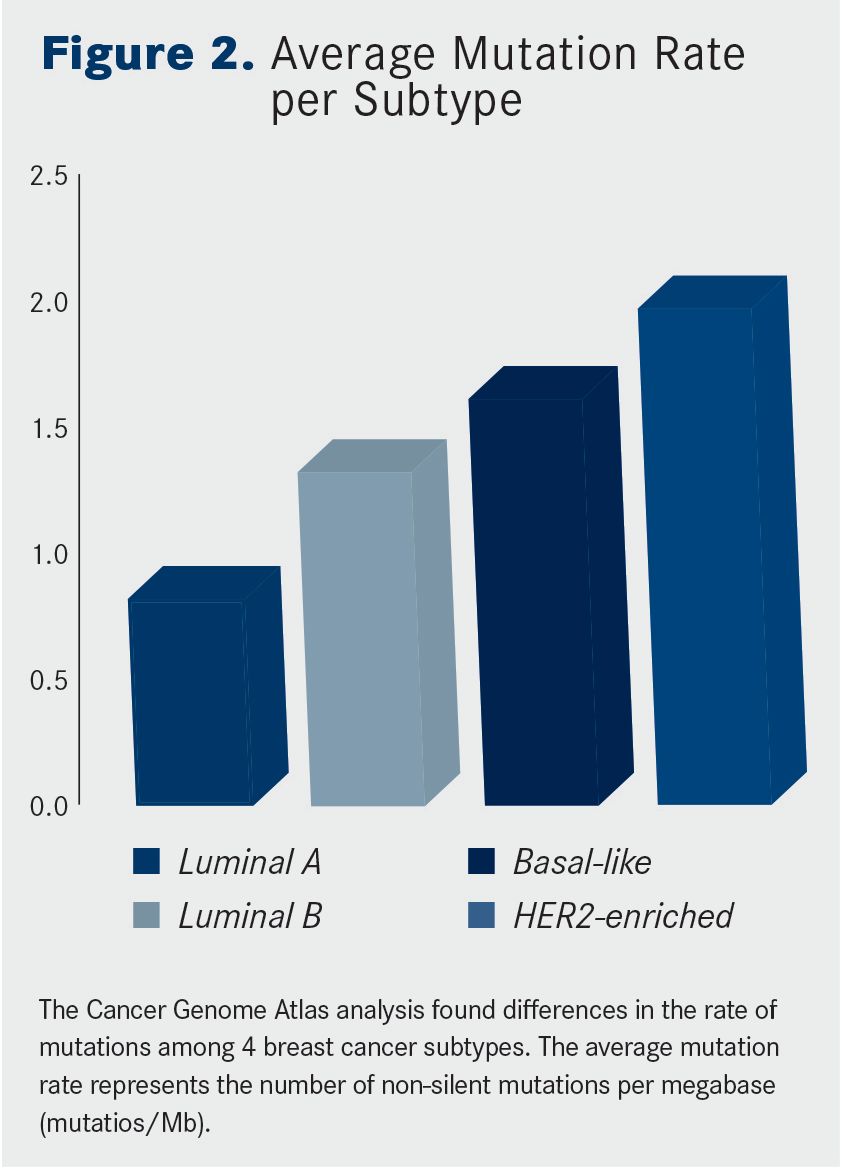
One of the most comprehensive sequencing studies was performed by The Cancer Genome Atlas (TCGA) research network. Dramatic differences in both the type and frequency of mutations across the clinical breast cancer subtypes were observed (Figures 1 and 2.)
Luminal Tumors
Overall, across 510 primary tumors that underwent whole-exome sequencing, 35 significantly mutated genes were identified. Many of these genes have been previously implicated in breast cancer, including components of the PI3K pathway, such as PIK3CA, PTEN, and AKT1, and the guardian of the genome, TP53. However, several of the mutated gene findings were novel, including TBX3, RUNX1, CBFB, AFF2, and CCND3.Luminal A tumors had the highest number of recurrently mutated genes, with the most frequent being the PIK3CA gene (45%). Other common mutations included MAP3K1, GATA3, CDH1, and MAP2K4. TP53 was also mutated in luminal A tumors.
The MAP2K4 and MAP3K1 genes are both involved in the JNK signaling pathway. This cascade, mediated by the Jun kinases, is involved in a myriad of cellular processes, including the response to stress.
Together, the 2 genes were mutated at a frequency of 20% in luminal A tumors; these mutations have been observed in several other sequencing studies, potentially implicating this pathway in breast cancer development.
Interestingly, luminal B tumors have a different mutation profile, suggesting another potential determinant of A versus B status. Luminal B tumors had a higher frequency of TP53 mutations, but fewer mutations in PIK3CA and the PI3K helical domain E545K. PIK3CA mutations are among the most common aberrations in breast cancer.
HER2-Enriched
The GATA3 gene was mutated at a similar rate in both types of luminal tumors, but the types of mutations differed; in the luminal A subtype they were observed mostly in intron 4, while in the luminal B subtype they occurred predominantly in exon 5.From a mutational perspective, in the HER2-enriched subtype, the most significantly mutated gene was TP53 and there were few other recurrently mutated genes.
Unsurprisingly, this subtype demonstrated frequent amplification of HER2 (80%). Somewhat more surprising was that not all HER2-positive tumors fell into the HER2-enriched clinical subtype—only approximately half did, while the rest were observed mostly in the luminal subtypes.
Furthermore, there were differences in the gene expression profiles of HER2-positive tumors that fell into the different clinical subtypes.
HER2-positive tumors within the HER2-enriched subtypes had significantly higher expression of a number of receptor tyrosine kinases, including EGFR and FGFR4, and they tended to be ER-negative, while those in the luminal subtype had lower levels of protein-based signaling and were more likely to be ER-positive.
Basal-Like Tumors
Another interesting finding was that, although all 4 subtypes had recurrent mutations in the PIK3CA gene, luminal tumors lacked other markers of PI3K pathway activation, such as increased levels of phosphorylated AKT, S6, and 4EBP1 proteins, biomarkers that were present in HER2-enriched and basal-like tumors.When broken down by subtype, luminal A tumors had the lowest overall mutation rate and the basal-like and HER2-enriched subtypes had the highest.
Interestingly, the situation was reversed when it came to the number of significantly mutated genes; luminal A and B tumors had the most, while basal-like and HER2-enriched tumors had only a handful each.
These findings reflect the fact that basal-like breast cancers are the most heterogeneous; they have more mutations, but few are recurrently mutated. This subtype was dominated by mutations in the TP53 gene, observed in 80% of samples, which in part helps, in part, to explain this genomic diversity, since the tumor suppressor protein p53 plays such an integral role in regulating the stability of the genome.
The type of TP53 mutation differed from those observed in luminal tumors. In basal-like tumors, there was a predominance of nonsense and frame-shift mutations compared with mostly missense mutations in luminal tumors.
PIK3CA and RB1 were also significantly mutated in basal-like tumors.
Notably, the breast cancer susceptibility genes BRCA1 and BRCA2 were among the deleterious germline variants identified in the TCGA samples and were enriched in patients with basal-like tumors.
Shining a Light on the Whole Genome
In fact, genomic alterations in basal-like tumors were similar to those found in high-grade serous ovarian tumors, a cancer type in which patients with BRCA1/2 mutations now have approved an therapeutic options in the form of a poly(ADP)-ribose polymerase (PARP) inhibitor. In addition to germline and/or somatic variants in the BRCA1/2 genes found in around 20% of basal-like tumors, the BRCA1/2 genes were also frequently deleted and hypermethylated.The sequencing studies performed to date have mostly been focused on the protein-coding regions of the genome. More recently, the first comprehensive whole-genome sequencing study was performed by an international group of researchers, led by the Sanger Institute in the United Kingdom.
In an effort to provide a more complete analysis of the breast cancer genome, including mutations that might be found within the nonprotein coding regulatory regions and the potential role of gene rearrangements in driving this disease, the researchers performed whole-genome sequencing on 560 tumor samples.
The study confirmed the substantial mutational heterogeneity seen in whole-exome analyses: likely driver mutations were discovered in 93 cancer genes, with at least 1 driver identifiable in 95% of cases. The 10 most commonly mutated genes were TP53, PIK3CA, MYC, CCND1, PTEN, HER2, FGFR1, GATA3, RB1, and MAP3K1. In ER-positive samples, PIK3CA was the dominant mutation, while TP53 mutations were more prevalent in ER-negative samples.
Although gene fusions and noncoding mutations as drivers of breast cancer were rare, analysis of the noncoding regions of the genome identified some areas with extremely high rates of mutation, although it was unclear if any of the mutated genes were drivers.
The authors suggested that these highly mutated areas resulted from a phenomenon known as kataegis, a pattern of localized hyper-mutation observed in nearly half of all samples in this study. It is thought that the APOBEC family of enzymes is responsible for kataegis in cancer cells. These enzymes are involved in RNA editing and may direct the frequent cytosine to thymine mutations observed in the kataegis process.
This study also examined the processes responsible for generating the somatic mutations observed in breast cancer—the so-called mutational signatures. They described 12 base-substitution signatures, 5 that had been outlined before in breast cancer and the remainder being novel.
They also uncovered 6 rearrangement signatures, 3 of which were associated with defective homologous recombination DNA repair mechanisms via loss of BRCA1, through loss of both BRCA1 and BRCA2, and by an as-yet-unknown mechanism.

Slow Progress in Clinical Translation
While these NGS studies were not specifically designed to be directly translated into clinical practice, the hope was that they would reveal potentially druggable targets to recapitulate the success of HER2-targeted therapies in breast cancer.
Unfortunately, the development of new therapies stemming from NGS findings has been hindered by the complex nature of the breast cancer genome. Nevertheless, important insights and potential new therapeutic strategies have been revealed to some extent.
Given that they are among the most frequently mutated genes across all breast cancer subtypes, PIK3CA and TP53 make ideal targets. As a kinase, PI3K is readily druggable and, indeed, PI3K inhibitors have become a prime therapeutic candidate.
Inhibitors of the mTOR protein, central to the downstream regulation of the PI3K pathway, have also been tested in breast cancer. In 2012, everolimus was approved by the FDA in combination with exemestanse, an aromatase inhibitor, for the treatment of advanced, ER-positive disease.
Clinical trials of PI3K inhibitors are ongoing, but have been complicated by the fact that, in many cases, PIK3CA mutation status does not predict benefit from treatment with these drugs. As yet, none have been approved in this disease setting.
On the other hand, p53 is not readily targetable. Research has instead focused on activated signaling molecules downstream of the tumor suppressor, including CHEK1/2, ATM, and ATR, or regulators of p53 function, such as MDM2. Although inhibitors of CHEK1/2, a protein that forms part of the DNA damage-signaling response pathway, reached clinical development, they proved mostly disappointing and only a single drug, LY2606368, continues to be evaluated (NCT02203513).
The similarities between high-grade serous ovarian cancer and basal-like tumors suggested that these tumor types may have common molecular drivers and could benefit from similar therapeutic strategies.
Indeed, the presence of BRCA1/2 mutations and other DNA repair defects, suggests a therapeutic opportunity for poly(ADP)-ribose polymerase (PARP) inhibition. Although this treatment strategy has not proved as effective in breast cancer as in ovarian cancer, numerous clinical trials are ongoing, including the phase III EMBRACA trial of talazoparib, which is expected to complete enrollment this year (NCT01945775).
Other exciting, clinically actionable findings from genome sequencing studies, include the discovery of HER2 mutations in patients who do not display HER2 amplification. This implies that HER2-targeted therapy could be more broadly applicable to patients with breast cancer.
A phase II trial of neratinib, which inhibits the HER2 receptor, is being conducted in patients with metastatic disease whose tumors are HER2-negative on immunohistochemistry or fluorescence in situ hybridization testing but harbor HER2 mutations (NCT01670877).
A recently published study used gene silencing techniques to identify the essential genes in a variety of breast cancer cell lines in an effort to find druggable targets and identified BRD4 as an essential gene in luminal breast cancer.
This gene encodes a BET bromodomain-containing protein that acts as an epigenetic “reader.” By binding to specific epigenetically modified regions of the genome and recruiting specific effector proteins, readers act as drivers of the transcriptional response.
Bromodomain-containing proteins have been shown to be critical to what is known as the adaptive bypass response, whereby cancer cells develop resistance to kinase inhibitors by upregulating kinases involved in alternative signaling pathways that make the targeted kinase redundant. BET inhibitors are beginning to be explored in the early stages of clinical development in solid tumors.

The Next Step: Proteogenomics
Researchers are now recognizing the need to move beyond genomics to answer further questions about the genes that are driving breast cancer.
The first large-scale proteogenomic study of breast cancer was recently published in Nature. The study authors, including members of the National Cancer Institute’s Clinical Proteomic Tumor Analysis Consortium, took advantage of advances in mass spectrometry-based proteomics to link the DNA mutations identified in the TCGA study to protein signaling.
The idea was to better understand how the genomic aberrations seen in breast cancer actually translate into cellular outcomes and thus determine which mutations are really essential tumor drivers and how they act in this way. Ultimately, the hope is to find more readily druggable targets and bridge the gap between cancer genomics and clinical applicability.
This kind of analysis can also help to clarify the role of potential molecular drivers present within large amplified or deleted regions of DNA. An example is the frequent loss of the long arm of chromosome 5 (5q) in basal-like breast cancers.
Proteomic analysis helped to identify potential drivers within this region by linking them to downstream protein signaling consequences. The study identified the loss of the CETN3 and SKP1 genes within this region and connected them to the upregulation of EGFR, already a known marker of basal-like tumors.
Jane de Lartigue, PhD, is a freelance medical writer and editor based in New Haven, Connecticut.
KEY RESEARCH
Ellsworth RE, Blackburn HL, Shriver CD, et al. Molecular heterogeneity in breast cancer: state of the science and implications for patient care [published online August 26, 2016]. Semin Cell Dev Biol. pii: S1084-9521(16)30266-X. doi:10.1016/j.semcdb.2016.08.025.
Goncalves R, Warner WA, Luo J, Ellis MJ. New concepts in breast cancer genomics and genetics. Breast Cancer Res. 2014;16(5):460-471. doi:10.1186/s13058-014-0460-4.
Marcotte R, Sayad A, Brown KR, et al. Functional genomic landscape of human breast cancer drivers, vulnerabilities, and resistance. Cell 2016;164(1-2):293-309. doi:10.1016/j.cell.2015.11.062.
Mertins P, Mani DR, Ruggles KV, et al. Proteogenomics connects somatic mutations to signalling in breast cancer. Nature. 2016;534(7605):55-62. doi: 10.1038/nature18003.
Nik-Zainal S, Davies H, Staaf J, et al. Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature. 2016;534(7605):47-54. doi:10.1038/nature17676.
Santarpia L, Bottai G, Kelly CM, et al. Deciphering and targeting oncogenic mutations and pathways in breast cancer. The Oncologist. 2016;21(9):1063-1078. doi:10.1634/theoncologist.2015-0369.
The Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumors. Nature. 2012;490(7418):61-70. doi:10.1038/ nature11412.
Verigos J, Magklara A. Revealing the complexity of breast cancer by next-generation sequencing. Cancers. 2015;7(4):2183-2200. doi:10.3390/cancers7040885.





























